Rを用いたRNA-seq結果に基づくKEGG解析

- アウトライン
-
- 作成日: 2026/01/14
- 更新日: –
はじめに
ニュートリゲノミクスは、栄養素や食事成分が遺伝子発現や分子ネットワークに及ぼす影響を網羅的に理解することを目的とした研究分野です。
食事因子は単一の遺伝子に作用するのみならず、代謝、炎症、シグナル伝達など複数の生物学的経路に同時に影響を及ぼすことが知られており、これらの影響を体系的に解釈するための解析手法が求められます。
Kyoto Encyclopedia of Genes and Genomes (KEGG) 解析は、このような背景のもと、遺伝子発現変動を個々の遺伝子レベルではなく、機能的な経路 (パスウェイ) レベルで評価することを可能にする有用な手法です。
KEGGデータベースには、糖・脂質・アミノ酸代謝、インスリンシグナル伝達、免疫応答、酸化ストレス応答など、栄養と密接に関連する多様なパスウェイが体系的に整理されています。
KEGG解析を用いることで、栄養介入や食事条件によって変動した遺伝子群が、どの代謝経路やシグナル経路に有意に集積しているかを統計学的に明らかにすることが可能となります。したがって、KEGG解析は、栄養素が生体機能に及ぼす分子機構を包括的に理解し、ニュートリゲノミクス研究における仮説生成や作用機序の解明を支える重要な解析基盤として位置づけられています。
問題を始める前に
2.1 DEGsとは?
差次的発現解析の結果、条件間で統計的に有意な発現変動(増加または減少)を示した遺伝子は、差次的発現遺伝子 (Differentially Expressed Genes; DEGs) と呼ばれます。
DEGsの判定には、主に以下の指標が用いられます。
-
log2 Fold Change (log2FC)
条件間における発現量の変化の大きさを表す指標であり、正の値は発現の増加、負の値は発現の減少を示します。
-
調整済み p 値 (adjusted p-value)
多数の遺伝子を同時に検定することによる多重検定の影響を補正した p 値で一般に False Discovery Rate (FDR) に基づく補正が用いられ、padjとして表記されることが多いです。
これらの指標をもとに、例えば padj<0.05などの基準を満たす遺伝子を、差次的発現が認められた遺伝子、すなわち DEGsとして取り扱います。
2.2 KEGG解析とは?
KEGG解析 (Kyoto Encyclopedia of Genes and Genomes 解析) は、RNA-seq やマイクロアレイ解析などによって得られた差次的発現遺伝子 (DEGs) リストを基に、特定の生物学的経路(パスウェイ)が統計学的に有意に富化しているかを評価する手法です。
KEGGデータベースには、代謝経路、シグナル伝達経路、細胞周期、免疫応答、疾患関連経路などが体系的に整理されており、個々の遺伝子変動を機能的な文脈で解釈することが可能となります。
一般的にKEGG解析では、入力した遺伝子群が背景遺伝子集合と比較して、特定のパスウェイにどの程度多く含まれているかを、過剰代表解析 (over-representation analysis) によって評価します。多数のパスウェイを同時に検定するため、Benjamini–Hochberg 法などによる多重検定補正を行い、偽発見率(FDR)を制御した補正後 p 値に基づいて有意な経路を同定します。
このように KEGG 解析は、網羅的な遺伝子発現データを生物学的機構へと統合し、実験条件によって影響を受ける主要な分子経路を明らかにするための重要な解析手法です。
2.3 ENTREZ IDとは?
ENTREZ ID は、NCBI (National Center for Biotechnology Information) が管理する遺伝子の一意な識別子であり、各遺伝子に対して重複しない番号が付与されています。
遺伝子名 (SYMBOL) は別名や表記揺れが存在するのに対し、ENTREZ ID は解析上の曖昧さがなく、データベース間での情報統合に適している点が特徴です。GO 解析や KEGG 解析など多くの機能解析ツールでは、標準的な入力形式として ENTREZ ID が用いられており、遺伝子注釈情報との対応付けが容易となります。
そのため、RNA-seqなどで得られた Ensembl IDや遺伝子シンボルを、解析前にENTREZ ID に変換することが一般的であり、信頼性の高い機能解析を行う上で重要な前処理ステップとなります。
問題
RNA-seq のカウントデータおよび各サンプルの処理条件(dex)を読み込み、遺伝子 × サンプルのカウント行列とサンプル情報を確認した上で、DESeq2 を用いて差次的発現解析を行ってください。
解析にあたっては、デザイン式に処理条件(dex)を用い、コントロール群(untrt)を基準としてモデルを構築してください。
次に、多重比較補正後の p 値(False Discovery Rate; FDR)を用いて padj < 0.05 を満たす遺伝子を差次的発現遺伝子(DEGs)として抽出し、ヒトの遺伝子注釈データベースを用いて KEGG 解析を行ってください。
さらに、得られた解析結果をもとにドットプロットを作成し、統計的に有意な KEGG パスウェイのうち上位 20 個を可視化してください。
【データ】
- Bioconductor パッケージ airway に含まれる RNA-seq データを使用する
-
本データは、デキサメタゾン (dex) 処理の有無による遺伝子発現の違いを測定したものである
- dex = trt: 処理群
- dex = untrt: コントロール群
解答
#サンプルデータの読み込み
library(airway)
data("airway")
# カウント行列 (遺伝子 × サンプル)
count_data <- assay(airway)
# サンプル情報 (投与 / 非投与)
col_data <- as.data.frame(colData(airway))
col_data$dex <- relevel(col_data$dex, "untrt")
head(count_data[, 1:3])
head(col_data)
# DESeq2のモデル構築
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = count_data,
colData = col_data,
design = ~ dex)
dds <- DESeq(dds)
res <- results(dds)
head(res)
# 有意な差次的発現遺伝子 (DEGs) の抽出
#5% FDRで抽出
sig <- res[res$padj < 0.05 & !is.na(res$padj), ]
head(sig[order(sig$log2FoldChange, decreasing = TRUE), ])
# log2FC 高い順に並べ替え (任意)
sig <- sig[order(sig$log2FoldChange, decreasing = TRUE), ]
# 差次的発現遺伝子 (DEGs) の出力
write.csv(sig, file = "DEG_sig.csv", row.names = TRUE)
# KEGG解析用パッケージの読み込み
library(clusterProfiler)
library(org.Hs.eg.db)
# Ensembl IDの抽出
symbols <- rownames(sig)
# ENTREZ IDへの変換
gene_df <- bitr(
symbols,
fromType = "ENSEMBL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
# ENTREZ IDの抽出
gene_ids <- gene_df$ENTREZID
# KEGG解析
ekegg <- enrichKEGG(
gene = gene_ids,
organism = "hsa", # ヒト (マウス: mmu)
pAdjustMethod = "BH",
pvalueCutoff = 0.05)
head(as.data.frame(ekegg))
# KEGG解析結果の可視化
library(enrichplot)
dotplot(ekegg, showCategory = 20)解説
※サンプルデータの読み込みから差次的発現遺伝子(DEGs)の抽出までの一連の流れについては、「RNA-seq解析をブラックボックスにしないために」にて詳しく解説しています。本節では、その後の KEGG 解析に焦点を当てて説明します。

5.1 差次的発現遺伝子 (DEGs) の出力
write.csv(sig, file = "DEG_sig.csv", row.names = TRUE)解析によって得られた差次的発現遺伝子 (DEGs) をcsv形式のファイルで保存します。
write.csv() は、Rのデータ (データフレームや行列) をcsvファイルとして書き出すためのコマンドです。保存したcsvファイルには、遺伝子IDや発現変化量 (log2 Fold Change)、p値などが出力されます。
5.2 KEGG解析用パッケージの読み込み
library(clusterProfiler)
library(org.Hs.eg.db)clusterProfilerパッケージは遺伝子リストを用いて体系的に機能解析を行うためのBioconductorパッケージであり、このパッケージを読み込むことでKEGG解析を行うことができるようになります。
org.Hs.eg.dbはKEGG解析を実施する際に用いるヒトの遺伝子注釈データベースのパッケージです。ヒト (org.Hs.eg.db) 以外にも、マウス (org.Mm.eg.db)やショウジョウバエ (org.Dm.eg.db) など多様な生物種のデータベースを指定することができます。
5.3 Ensembl ID の抽出
symbols <- rownames(sig)出力したcsvファイルでは、差次的発現遺伝子のEnsembl IDが抽出されています。しかし、KEGG データベースおよびclusterProfilerのenrichKEGG関数では、標準的に ENTREZ IDが用いられるため、Ensembl IDをENTREZ ID に変換する必要があります。
rownames() はデータフレームの行名を抽出するコマンドです。出力したcsvファイルから行名であるEnsembl IDを抽出し、"symbols" として保存します。
5.4 ENTREZ IDへの変換
gene_df <- bitr(symbols, fromType = "ENSEMBL", toType = "ENTREZID",
OrgDb = org.Hs.eg.db)bitr() は、clusterProfilerパッケージに含まれている遺伝子ID同士を相互変換 (IDマッピング) するための関数です。fromTypeに変換前の遺伝子ID (=”Ensembl”)、toTypeに変換後の遺伝子ID (=”ENTREZ ID”) を指定します。また、変換を行う際に参照する生物種のDBをorgDbに指定します。今回はヒトのDB“org.Hs.eg.db”を用います。変換した結果は”gene_df”として保存されます。
5.5 ENTREZ ID の抽出
gene_ids <- gene_df$ENTREZID変換したENTREZ IDを抽出し”gene_ids”として保存します。抽出したENTREZ IDを用いて、この後のKEGG解析を行います。
5.6 KEGG解析
ekegg <- enrichKEGG(gene = gene_ids, organism = "hsa", # ヒト (マウス:
mmu) pAdjustMethod = "BH", pvalueCutoff = 0.05)enrichKEGG() は、差次的発現遺伝子を用いて、生物学的に意味のある経路(Pathway) を見つけるための関数です。"gene"に解析に用いる遺伝子 (gene_ids) を指定します。"organism"にはKEGG解析で用いる生物種コードを指定します。KEGG解析では、遺伝子ID、パスウェイ構造が生物ごとに異なるため、「この遺伝子リストはどの生物のものか?」を明示する必要があります。今回の解析では、ヒト遺伝子 (“hsa”) を指定します。
KEGG解析を実施するにあたり、多重検定問題を考慮するため、Benjamini–Hochberg 法を用い、FDR < 0.05 を有意とします。KEGG解析の結果は"ekegg"として保存されます。
5.7 データの確認
head(as.data.frame(ekegg))head() はKEGG解析結果の先頭数行を表示するコマンドです。KEGG解析が適切に実施されているかを確認することができます。
5.8 KEGG解析結果の可視化
library(enrichplot)
dotplot(ekegg, showCategory = 20)library() は、KEGGの解析結果を「図として可視化するためのパッケージ」を読み込むためのコマンドです。
dotplot() は、解析結果をドットプロットとして可視化するコマンドです。dotplot (ekegg, showCategory = 20) と指定することで、KEGG解析の結果から「有意な上位20個の代謝経路」をドットプロットとして可視化します。
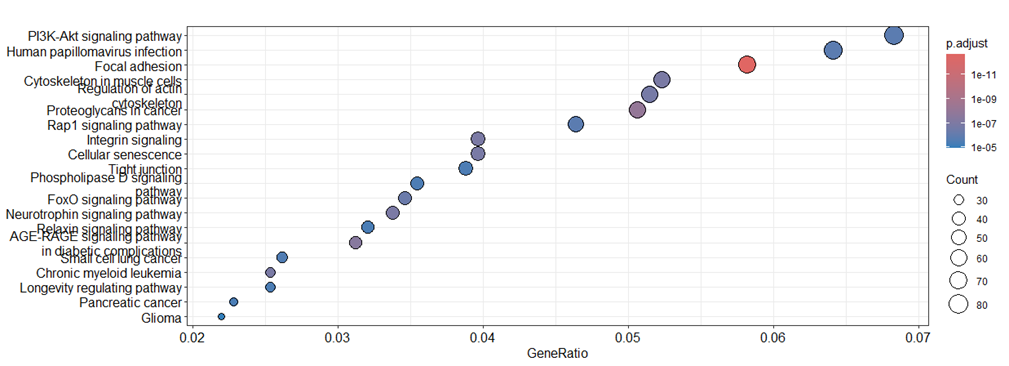
図1: ドットプロット
ドットプロットは横軸がGeneRatio、点の大きさがヒット遺伝子数を示しています。今回の結果から、”PI3K-Akt signaling pathway”に関連する遺伝子が最も多くヒットしていることが示唆されました。PI3K-Akt signaling pathwayは、細胞増殖・生存・アポトーシス抑制を制御する主要なシグナル伝達経路であり、がん化や細胞応答において重要な役割を果たすことが知られています。
まとめ
本稿では、デキサメタゾン (dex) 処理の有無による遺伝子発現変動を解析するため、airwayデータセットを用いたRNA-seq解析を行いました。DESeq2による差次的発現解析の結果、FDR < 0.05を満たす差次的発現遺伝子 (DEGs) を多数同定しました。
これらのDEGsを対象としてKEGG解析を実施したところ、複数の生物学的経路が有意に富化していることが示唆されました。特に、「PI3K-Akt signaling pathway」はGeneRatioおよびヒット遺伝子数の観点から最も顕著な経路として同定されました。
PI3K-Aktシグナル伝達経路は、細胞増殖、生存、アポトーシス抑制を制御する中心的な経路であり、がん化、ストレス応答、代謝調節など多様な細胞応答に関与しています。本解析結果は、デキサメタゾン処理が細胞の生存・増殖に関わるシグナルネットワークへ影響を及ぼす可能性を示唆するものであり、栄養・環境因子による分子機構理解において KEGG 解析が有用であることを示す一例と考えられます。
ただし、本解析は過剰代表解析に基づく探索的解析であり、個々の遺伝子の因果的関与を直接示すものではない点には留意が必要です。
