Rによる差次的タンパク質解析

- アウトライン
-
- 作成日: 2026/01/18
- 更新日: –
はじめに
近年、LC–MS/MSによる定量プロテオーム解析は、基礎研究から臨床研究、さらには栄養学・ニュートリゲノミクス分野に至るまで幅広く利用されています。一方で、解析工程の多くが専用ソフトウェアや自動化パイプラインに依存しているため、「どのような前提のもとでデータが処理され、どの統計モデルによって差が評価されているのか」が利用者から見えにくいという課題があります。
特に、欠測値を多く含むプロテオームデータでは、解析手法の選択や前処理の違いが結果の解釈に大きな影響を及ぼします。しかし、こうした点が明示されないまま結果のみが提示されることも少なくありません。
本コラムでは、公開されているプロテオームデータセットを用い、R による差次的タンパク質解析の最小構成スクリプトを示します。あえて高度な拡張や自動化は行わず、
- どのデータを使い
- どの前提で正規化を行い
- どの統計モデルで群間差を評価しているのか
を一つ一つ確認できる形で解析を進めます。これにより、プロテオーム解析をブラックボックスとして扱うのではなく、結果を自ら解釈できる基盤を身につけることを目的とします。
問題を始める前に
2.1 問題設定
以下の条件を満たす解析を考えます。
- Controlを基準とした複数条件との比較
- Label-free 定量プロテオーム
- 正規化前の定量データを用い、解析内で正規化を行う
- 統計解析は線形モデルに基づく方法
- 有意なタンパク質を可視化する
2.2 使用する公開データ
本演習では、RパッケージDEPに付属する公開プロテオームデータを使用します。
問題
次の問いに答えなさい。
- プロテオーム定量データをRに読み込め
- 群 (Control / Treatment) 情報を用いて差次的タンパク質解析を行え
- 有意なタンパク質を抽出し、Volcano plot を作成せよ
解答
# 初回のみ
# install.packages("BiocManager")
# BiocManager::install("DEP")
library(DEP)
# データ読み込み
data("DiUbi")
data("DiUbi_ExpDesign")
# タンパク質IDの一意化
DiUbi_unique <- make_unique(
DiUbi,
"Majority.protein.IDs",
"Gene.names"
)
# LFQ intensity 列を抽出
lfq_cols <- grep("^LFQ.intensity", colnames(DiUbi_unique))
# SummarizedExperiment作成
se <- make_se(
DiUbi_unique,
DiUbi_ExpDesign,
columns = lfq_cols
)
# 正規化
se_norm <- normalize_vsn(se)
# 差次的解析
se_diff <- test_diff(
se_norm,
type = "control",
control = "ctrl"
)
# 有意判定を追加
se_diff <- add_rejections(
se_diff,
alpha = 0.05
)
# 結果抽出
res <- get_results(se_diff)
head(res)
# Volcano plot
plot_volcano(
se_diff,
contrast = "K11_vs_ctrl",
label_size = 3
)解説
5.1 パッケージ読み込み
# 初回のみ
# install.packages("BiocManager")
# BiocManager::install("DEP")
library(DEP)本解析では、Bioconductor パッケージ DEPを使用しました。
DEPは、プロテオーム解析で必要となる・・・
- データ前処理
- 正規化
- 差次的タンパク質解析
- 可視化
を一貫して扱えるパッケージです。
install.packages() や BiocManager::install() は 初回セットアップ時のみ実行してください。
5.2 サンプルデータの読み込み
data("DiUbi")
data("DiUbi_ExpDesign")DEP に付属する UbIA-MS (ユビキチン相互作用) プロテオームデータを読み込みます。
-
DiUbi
タンパク質定量データ (行=タンパク質、列=サンプル)
-
DiUbi_ExpDesign
実験デザイン情報 (条件、リプリケート)
これは RNA-seq解析で言うcount matrix + colDataに相当する構造です。
5.3 タンパク質IDの一意化
DiUbi_unique <- make_unique(
DiUbi,
"Majority.protein.IDs",
"Gene.names"
)プロテオームデータでは、1つの遺伝子やタンパク質に複数の同定IDが対応することがあります。しかし、統計解析では各行が一意な単位であることが必須です。
make_unique() を使用して、
- Majority.protein.IDsを解析用の一意ID
- Gene.namesを表示用ラベル
として、重複があれば自動的に番号を付与し、一意な行名を作成しました。
5.4 定量値の選択
lfq_cols <- grep("^LFQ.intensity", colnames(DiUbi_unique))MaxQuant 由来のプロテオームデータには、複数の定量指標が含まれます。
- iBAQ: 絶対量推定向き
- LFQ intensity: 群間比較向き
本例では 差次的解析を目的としているため、LFQ.intensity 列のみを解析対象としました。
5.5 SummarizedExperimentの作成
se <- make_se(
DiUbi_unique,
DiUbi_ExpDesign,
columns = lfq_cols
)DEPは、Bioconductor標準クラスSummarizedExperimentを用いて解析を行いました。
このオブジェクトには・・・
- 定量データ (assay)
- タンパク質注釈 (rowData)
- サンプル情報 (colData)
が安全に一体化して格納されます。
5.6 正規化
se_norm <- normalize_vsn(se)サンプル間の分布差を補正しています。MSデータでは、測定感度、注入量、機器ドリフトなどにより、サンプル間で全体強度がずれるためです。
この操作により、強度依存的な分散を安定化し線形モデルの前提 (等分散性) に近づけます。
5.7 差次的タンパク質解析
se_diff <- test_diff(
se_norm,
type = "control",
control = "ctrl"
)各タンパク質についてTreatmentとControlの群間差を推定します。
5.8 有意判定の追加
se_diff <- add_rejections(
se_diff,
alpha = 0.05
)test_diff() ではp値が計算されますが、「有意かどうか」の判定基準はまだ定義されていません。
add_rejections() により、FDR < 0.05 を有意と定義し。各比較に対するsignificantフラグを追加しました。
5.9 結果の抽出
res <- get_results(se_diff)
head(res)差次的解析の結果を1つの表として取得します。この表には、各比較のp値・調整済みp値、log2 fold change、有意判定フラグがまとめて含まれます。
5.10 Volcano plot による可視化
plot_volcano(
se_diff,
contrast = "K11_vs_ctrl",
label_size = 3
)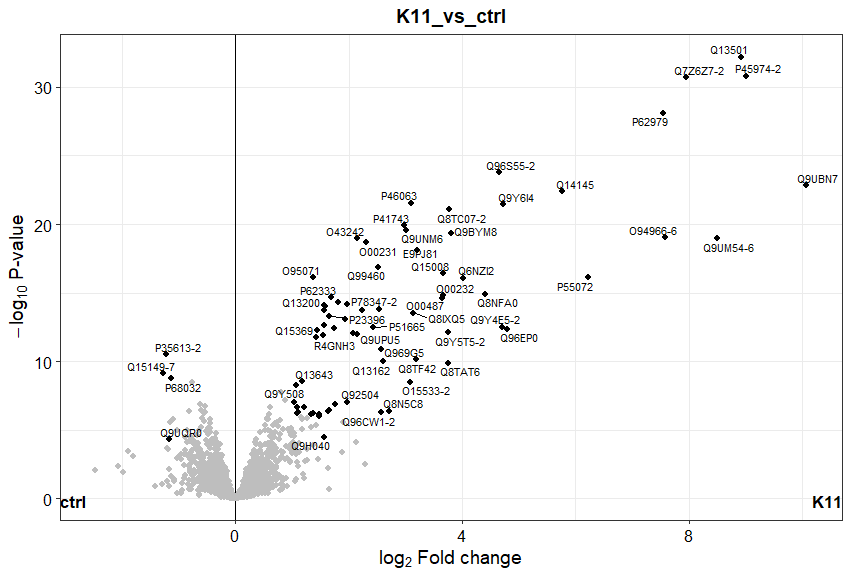
Volcano plot は、
横軸: log2 fold change
縦軸: −log10(p 値)
を用いて、効果量と有意性を同時に可視化する図です。
欠測値を含むタンパク質は自動的に除外されるため、図に表示されるのは統計的に比較可能だったタンパク質のみです。
まとめ
本コラムでは、公開プロテオームデータを用いて、Rによる差次的タンパク質解析の最小構成を示しました。プロテオーム解析では欠測値を含むことが一般的であり、すべてのタンパク質について群間差が推定できるとは限りません。
本解析では、そうした実データの特性を前提とした上で、正規化、統計解析、有意判定、可視化までを明示的に記述しました。
Rを用いることで、解析手法の前提や限界をコードとして明確に残すことができる点は、大きな利点です。探索的解析であっても、どのような仮定のもとで得られた結果なのかを理解しておくことは、後続の検証研究や解釈の妥当性を高める上で重要です。
本稿が、プロテオーム解析をブラックボックスとして扱わず、自ら結果を読み解くための出発点となれば幸いです。
