DNAマイクロアレイ解析における正規化の考え方と実践

- アウトライン
-
- 作成日: 2026/1/17
- 更新日: –
はじめに
生物の設計図とも例えられるDNAには、細胞の性質や振る舞いを規定する多数の遺伝子情報が記されています。しかし、これらの遺伝子が常に一様に働いているわけではありません。細胞は、置かれた環境や生理的状態に応じて、必要な遺伝子を選択的に利用し、その発現量を精密に制御しています。
DNAに記された遺伝情報が読み取られ、機能性分子の合成へとつながる一連の過程は「遺伝子発現」と呼ばれます。その最初の段階が、DNAの塩基配列情報がRNAに写し取られる「転写」です。転写は、遺伝子発現の出発点として極めて重要なプロセスです。
転写によって生成されるRNAには複数の種類がありますが、その中でタンパク質合成の設計図として機能するのがmRNA (メッセンジャーRNA) です。細胞内に存在するmRNAの量は、対応する遺伝子がどの程度活発に利用されているかを反映しているため、mRNA量を測定することで遺伝子発現状態を定量的に把握することが可能になります。
このようなmRNA量を多数の遺伝子について同時に測定する手法が「DNAマイクロアレイ解析」です。本手法では、既知の遺伝子配列をもとに設計されたDNA断片 (プローブ) を基板上に固定し、細胞から抽出・標識したmRNA (あるいはそれを鋳型として合成したcDNA) を結合させます。各プローブに結合した分子量をシグナルとして読み取ることで、数千から数万遺伝子の発現量を一括して解析できます。
DNAマイクロアレイ解析は、処理条件の違いや疾患の有無など、条件の異なるサンプル間で遺伝子発現を比較する用途に特に適しています。細胞が外部刺激にどのように応答しているのか、あるいは状態変化に伴ってどの遺伝子が活性化・抑制されているのかを網羅的に把握できる手法として、これまで幅広い分野で利用されてきました。
問題を始める前に
2.1 DNAマイクロアレイ解析とは?
DNAマイクロアレイ解析は、細胞や組織から抽出したRNAを手がかりとして、遺伝子がどの程度発現しているかを多数同時に調べるための手法です。遺伝子発現解析の分野では、比較的早い段階から利用されてきた代表的な技術の一つとして知られています。
DNAマイクロアレイでは、ガラスなどの基板上に多数のDNAプローブが高密度に固定・整列されています。これらのプローブは、解析対象とする既知の遺伝子配列をもとにあらかじめ設計されたものです。試料中のRNAは逆転写によって cDNA に変換され、プローブと相補的な配列同士が結合する性質 (ハイブリダイゼーション) を利用して検出されます。結合量は蛍光シグナルとして読み取られ、その強度から各遺伝子の発現量が推定されます。
DNAマイクロアレイの大きな特徴は、数万種類にも及ぶ遺伝子の発現状態を1回の実験で網羅的に評価できる点にあります。一方で、解析可能なのはプローブが用意されている既知の遺伝子に限られます。そのため、RNA-seqのように未知の転写産物や新規スプライシングバリアントを探索することは、原理的に困難です。
しかし、この手法は長年にわたる利用実績があり、解析パイプラインや結果解釈の枠組みが比較的確立しているという利点を有しています。実験コストやデータ量もRNA-seqと比べて抑えやすく、条件間での遺伝子発現変化を比較する目的であれば、現在でも十分に実用的な選択肢といえます。
特に、処理群と対照群、あるいは疾患群と健常群といったグループ間で、「どの遺伝子の発現が増加または減少したのか」を比較する解析において、DNAマイクロアレイはその強みを発揮してきました。発現量の絶対値を厳密に求めるというよりも、相対的な変動に着目した解析に適した手法だと位置づけられます。
近年はRNA-seqが主流となりつつありますが、DNAマイクロアレイはトランスクリプトミクス研究の基盤を築いてきた重要な技術の一つです。これまでに蓄積された知見とデータ資産を踏まえると、研究目的、予算、必要とされる解析の深さに応じて、RNA-seqと適切に使い分けることが重要であると考えられます。
2.2 DNAマイクロアレイの種類
遺伝子発現解析に用いられるDNAマイクロアレイは、測定方法の違いにより、2色マイクロアレイと1色マイクロアレイの大きく2種類に分類されます。
これらは、サンプルの標識方法や得られるデータの性質、必要とされる前処理・正規化手法が異なります。
また、同じ1色法・2色法であっても、使用されるプラットフォーム (メーカーやアレイ設計) によって、プローブ構造や前処理手順が異なる点にも注意が必要です。
2.2.1 2色マイクロアレイ (cDNAマイクロアレイ、Stanford型、Agilent など)
2色マイクロアレイでは、2つのサンプルをそれぞれ異なる蛍光色素で標識し、同一アレイ上で同時に測定します。一般的にはCy3とCy5などの蛍光色素が用いられ、各スポットにおける蛍光強度比が、遺伝子発現量の相対的な差を表します。
この方式の最大の利点は、同一スライド上で2条件を直接比較できる点にあります。そのため、アレイ間のばらつきやスキャン条件の違いといった影響を受けにくく、条件間の相対比較を重視する実験デザインに適した手法とされています。
一方で、蛍光色素ごとの発光効率や退色特性の違いにより、色素間で系統的な強度差が生じやすいという課題があります。また、発現強度に依存して蛍光比が歪む「強度依存性バイアス」は、2 色マイクロアレイ特有の問題として知られています。
そのため、解析においては色素間の強度差や強度依存性を補正する正規化処理が不可欠です。代表的な手法として、局所回帰を用いて比の偏りを補正するLOWESS正規化が広く用いられています。
主なプラットフォーム
-
cDNA マイクロアレイ(Stanford 型)
初期の2色マイクロアレイとして広く用いられてきた形式で、比較的長いcDNA断片をプローブとしてガラススライド上にスポットします。高い感度を有する一方で、スポット品質やプローブ間ばらつきの影響を受けやすく、厳密な正規化および品質管理が重要となります。
-
Agilent マイクロアレイ
合成オリゴヌクレオチドを高精度に配置した2色マイクロアレイを提供する代表的なプラットフォームです。プローブ配列やスポット品質の再現性が高く、従来のcDNAマイクロアレイと比べて安定した測定が可能とされています。Agilent データでは、スポットごとの蛍光強度や比情報を用いた解析が一般的であり、LOWESS正規化を含む色素補正を前提とした前処理が重要となります。なお、Agilentマイクロアレイは従来2色法で広く利用されてきましたが、近年では1色法による測定も可能であり、用途に応じて使い分けられています
2.2.2 1色マイクロアレイ (GeneChipなど)
1色マイクロアレイでは、すべてのサンプルを同一の蛍光色素で標識し、1サンプルにつき1枚のアレイを用いて測定します。この方式では色素間差を考慮する必要がないため、データ構造が比較的単純になります。
一方で、異なるアレイ間で測定されたデータを比較するため、チップごとの測定条件やスキャン条件の違いが結果に影響しやすいという特徴があります。そのため、アレイ間の分布やスケールの違いを補正する正規化処理 (quantile normalizationなど) が重要となります。
主なプラットフォーム
-
Affymetrix(GeneChip)
GeneChipは、1つの遺伝子に対して複数の短いオリゴヌクレオチドプローブを配置する設計が特徴です。この構造により、個々のプローブのばらつきを統計的に統合することが可能となっています。解析では、背景補正・正規化・要約を一括で行う RMA (Robust Multi-array Average) などの専用前処理手法が広く用いられます。
-
Illumina BeadArray
Illumina BeadArrayも1色マイクロアレイに分類されます。このプラットフォームでは、同一配列のプローブがビーズ状に多数配置されており、高い測定再現性を有する点が特徴です。Illumina データでは、log2変換済みシグナルやdetection p-valueが提供されることが多く、解析時にはこれらの情報を用いたフィルタリングが行われる場合があります。
2.3 DNAマイクロアレイの公開データ
DNAマイクロアレイデータの多くは、現在では公共データベースを通じて広く公開されており、研究目的に応じて誰でも利用することができます。代表的なデータベースとして、以下の2つが挙げられます。
2.3.1 公共データベース
-
Gene Expression Omnibus (GEO)
GEOはNCBIが運営する、世界最大級の遺伝子発現データベースです。
1色・2色マイクロアレイをはじめ、さまざまなプラットフォームのデータが登録されています。R 環境ではGEOqueryパッケージを用いることで、GEOに登録されたデータを直接取得し、解析に利用することが可能です。
-
ArrayExpress
ArrayExpressはEMBL-EBIによって運営されている、GEOと並ぶ国際的な遺伝子発現データリポジトリです。BioconductorではArrayExpressパッケージなどを用いて、Rからデータにアクセスすることができます。
2.3.2 Rにおけるデータ構造
これらのデータベースから取得したマイクロアレイデータは、R上ではExpressionSetオブジェクトとして扱われることが一般的です。
GEO Matrix 形式のデータは、そのままExpressionSetとして読み込まれることが多く、生データから解析する場合はAffyBatchやRGListなどの形式を経て ExpressionSet に変換されます。
ExpressionSetには、
- 発現量の行列 (expression matrix)
- サンプル情報 (phenotype data)
- 実験条件や注釈情報 (metadata)
が一体として格納されており、その後の正規化や統計解析を効率的に行うことができます。
2.3.3 公開データの主な形式
公開されているマイクロアレイデータは、解析目的や提供形態の違いにより、主に以下の2種類に分類されます。
GEO Matrix形式
GEO Matrixは、すでに背景補正・正規化・要約処理などが施された発現行列データです。GEOqueryパッケージを用いることで、Rから直接取得し、すぐに解析を開始できるという利点があります。
一方で、どの正規化手法が用いられているかはデータ提供者に依存しており、解析者が前処理方法を自由に選択することはできません。そのため、探索的解析や全体傾向の把握には適していますが、前処理条件を厳密に制御した解析には不向きな場合があります。
生データ形式 (Raw data)
生データには、AffymetrixのCELファイルや、2色マイクロアレイにおけるスポット強度を記録したTXTファイルなどが含まれます。
これらのデータでは、背景補正や正規化といった前処理を解析者自身が行う必要があります。その分、実験デザインやデータ特性に応じて適切な前処理手法を選択できるという柔軟性があります。解析手順の再現性や妥当性を重視する研究では、生データからの解析が選択されることが一般的です。
2.3.4 GEO から生データを取得する手順
ここでは、1色マイクロアレイデータを例に、GEOから生データを取得する流れを確認します。
-
①GEO の検索ページにアクセスする
まず、NCBIが提供するGEOのトップページ (https://www.ncbi.nlm.nih.gov/geo/) にアクセスします。
GEOの検索画面では、キーワードやアクセッション番号を用いてデータを検索することができます。
論文や資料に”GSE2034“のようなIDが記載されている場合は、その番号を直接入力して検索します。
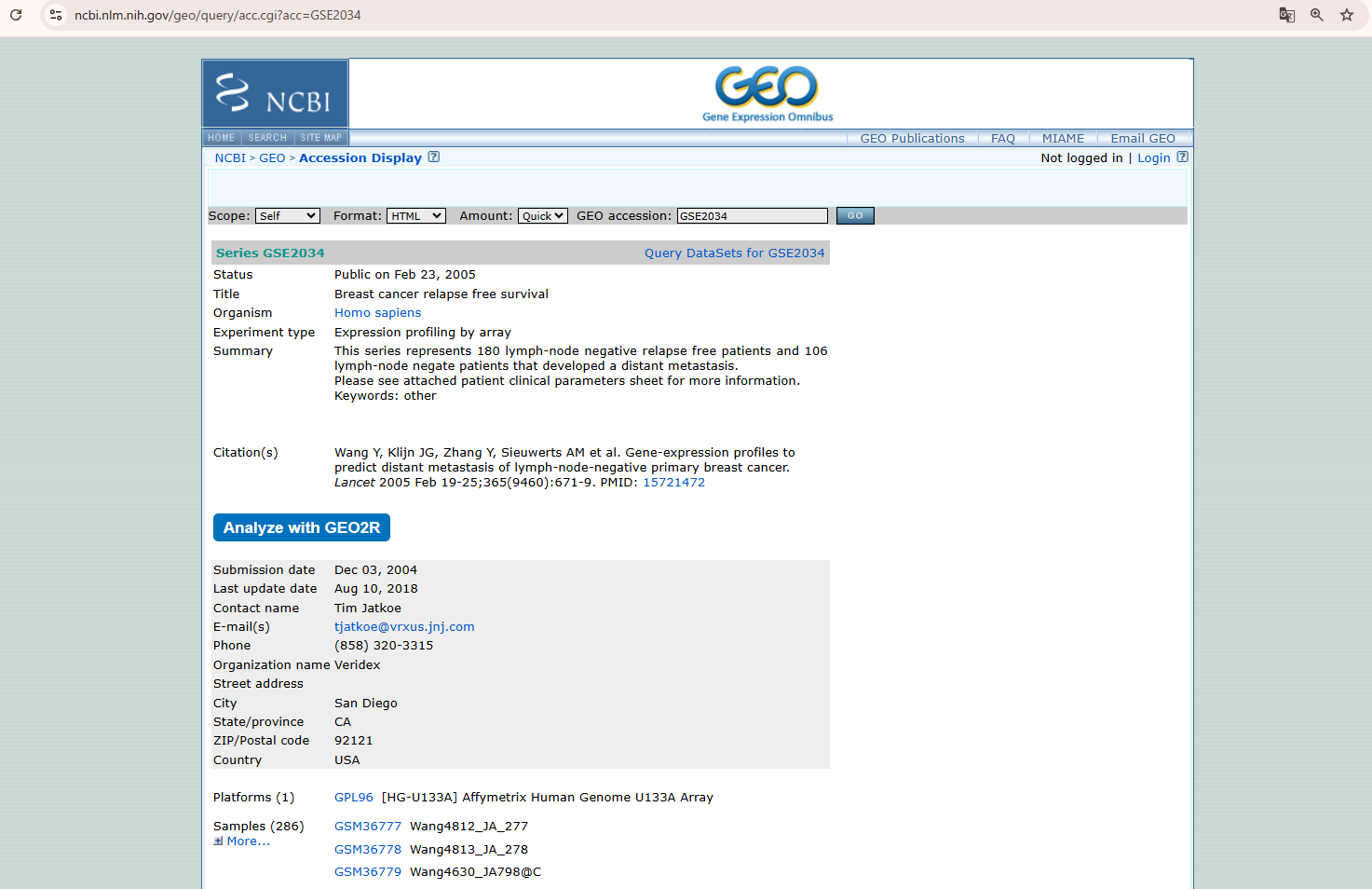
検索結果から、該当するGSE (Series) ページ を開きます。
GSEページには、以下のような情報がまとめて掲載されています。
- 研究の概要や目的
- 使用されたプラットフォーム (Affymetrix、Agilentなど)
- サンプル数や実験条件
- 論文情報
- 提供されているデータファイル
解析に使用する生データは、このページ内の”Supplementary iles“セクションから取得します。
-
②Supplementary filesから生データを探す
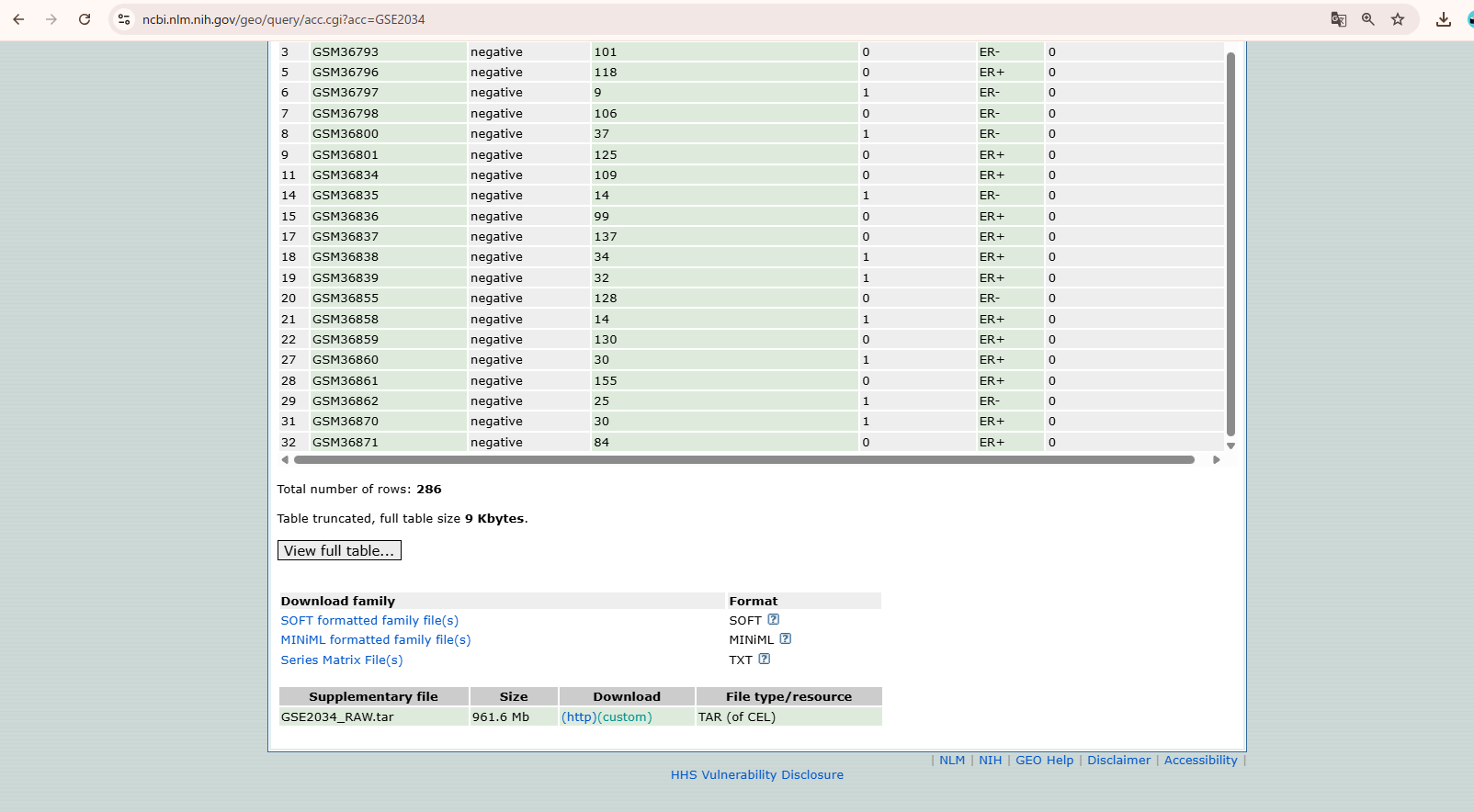
ページを下にスクロールすると、“Supplementary files (補足ファイル)”という項目があります。
ここには、以下のようなファイルが並んでいます。
- GSE2034_RAW.tar
- .CEL.gz
- .txt
- .gpr
Affymetrix の 1色マイクロアレイの場合は、CELファイル (または CEL を含むtar ファイル) が生データに該当します。
ファイル名に
- RAW
- CEL
といった文字が含まれているものが目印になります。
目的のファイル名をクリックすると、ブラウザ経由でダウンロードが開始されます。ダウンロード後はファイルを解凍することで、利用できます。
2.4 マイクロアレイデータに生じるばらつき
マイクロアレイ解析では、遺伝子ごとの発現量を蛍光強度として数値化します。しかし、得られるシグナルには、実験条件や技術的要因に由来するさまざまなばらつきが含まれます。これらのばらつきを考慮せずにデータを比較すると、遺伝子発現の差を過大または過小に評価し、誤った結論に至る可能性があります。
DNA マイクロアレイにおいて代表的に生じるばらつきは、以下のように整理できます。
2.4.1 蛍光色素の性質によるばらつき (色素間差)
DNAマイクロアレイ (特に2色マイクロアレイ) は、異なる蛍光色素で標識した cDNAを同一アレイ上で測定します。しかし、蛍光色素は同一条件下でも同じ強度で発光するとは限りません。
同量のmRNAが結合していたとしても、一方の色素が相対的に明るく、あるいは暗く測定されることがあり、これが偽陽性や偽陰性の原因となる場合があります。
2.4.2 技術的ばらつき
DNAマイクロアレイ測定には、多くの機器や操作工程が関与します。蛍光シグナルを読み取るスキャナーの設定差、ハイブリダイゼーション効率の違い、スライドの印刷品質や前処理条件のばらつきなどは、測定値に影響を与える代表的な技術的要因です。
これらの要因は、同一サンプルを測定しても異なる結果を生じさせる可能性があり、データの再現性や信頼性に直接関わります。
2.4.3 生物学的ばらつき
同一の実験条件下であっても、生物そのものの違いにより遺伝子発現には差が生じます。個体ごとの遺伝的背景、使用する組織や細胞の種類、さらにはサンプル採取時の生理状態や時間帯などが、発現パターンに影響を与えることがあります。
生物学的ばらつきは、実験データに本質的に含まれる変動であり、実験の再現性や結果解釈において重要な意味を持ちます。そのため、適切な実験デザインや十分なサンプル数の確保が不可欠です。
2.4.4 環境的ばらつき
実験を実施する環境条件も、マイクロアレイデータに影響を及ぼします。ハイブリダイゼーションや洗浄工程における温度・湿度の違い、操作担当者の手技のわずかな差などが、蛍光シグナルの強度や一貫性に影響する場合があります。
これらの環境的ばらつきは、実験条件の標準化や操作手順の統一によって、ある程度抑制することが可能です。
2.5 マイクロアレイデータの正規化
前節で述べたように、マイクロアレイデータには遺伝子発現そのものとは無関係な技術的・測定的ばらつきが含まれます。これらの影響を取り除き、異なるサンプル間で発現量を適切に比較できるようにする前処理が正規化 (normalization) です。
正規化の目的は、「生物学的差異を保持したまま、技術的バイアスのみを補正すること」
にあります。代表的な正規化手法を以下に示します。
-
全体強度の正規化 (Global normalization)
各スライドの蛍光強度の平均値や総和を基準として、すべてのスポット値をスケーリングする方法です。全体の発現量がサンプル間で大きく変化しないという前提のもとで用いられ、実装が簡便で計算コストが低いという利点があります。一方で、サンプル間で大規模な発現変動が存在する場合には、真の生物学的差異まで補正してしまう可能性があります。
-
中央値正規化 (Median normalization)
各スライドにおける蛍光強度の中央値を基準として補正を行う方法です。平均値に比べて外れ値の影響を受けにくく、極端なシグナルを含むデータに対しても比較的安定した補正が可能です。全体の発現分布が大きく偏っていないと仮定できる場合に有効とされます。
-
Lowess正規化 (Locally Weighted Scatterplot Smoothing)
主に2色マイクロアレイで用いられる正規化手法です。蛍光強度と発現比の関係に着目し、各スポットの強度に応じて局所的に回帰曲線を当てることで、色素間バイアスや強度依存性バイアスを補正します。非線形な補正が可能であり、色素特性の違いや非対称な分布を伴うデータにも柔軟に対応できる点が特徴です。
-
分位点正規化 (Quantile normalization)
複数のスライド間で、蛍光強度の分布そのものを一致させる方法です。各スライドのシグナルを順位付けし、同じ順位の値を平均化することで、全体の分布形状を統一します。大規模な比較解析や、アレイ間で発現分布が大きく異なる場合に特に有効ですが、「全サンプルで発現分布は概ね同じである」という仮定を暗に置く点には注意が必要です。
マイクロアレイの種類によって、正規化で補正すべき主なバイアスは異なります。
2色マイクロアレイ
→ 色素間の強度差・強度依存性の補正が主目的
1色マイクロアレイ
→ アレイ間の測定条件差を補正し、サンプル間比較を可能にすることが主目的
2.6 プラットフォームと正規化手法の対応関係
マイクロアレイ解析では、プラットフォームごとに主要なバイアスの性質が異なります。そのため、データの型 (1色・2色) やアレイ設計に応じて、適切な正規化手法を選択することが重要です。
以下に、代表的なプラットフォームと一般的に用いられる正規化手法の対応関係を示します。
2 色マイクロアレイでは、色素間バイアスおよび強度依存性バイアスの補正が解析の中心となります。
-
cDNA マイクロアレイ (Stanford型)
スポット品質やプローブ間ばらつきが比較的大きいため、Global normalizationやMedian normalizationに加え、LOWESS正規化による強度依存性バイアスの補正が重要となります。
-
Agilent マイクロアレイ (2色法)
プローブ設計やスポット再現性は高いものの、色素間の系統的差は依然として存在します。そのため、LOWESS 正規化を含む色素補正を前提とした解析が一般的に行われます。
これらの 2 色マイクロアレイでは、「発現比 (ratio) が歪まないように補正すること」が正規化の主目的となります。
問題
本問題では、R に用意されている公開DNAマイクロアレイデータを用いて、Affymetrix 1色マイクロアレイデータの正規化処理を実際に行うことを目的とします。
【データ】
- ① NCBI が提供する GEO のトップページhttps://www.ncbi.nlm.nih.gov/geo/にアクセスし、GSE2034 を検索する。
- ② GSE2034のSeries ページから、Supplementary filesに含まれるGSE2034_RAW.tar をダウンロードする。
- ③ ダウンロードした GSE2034_RAW.tarを解凍し、含まれている.CELファイルを、作業ディレクトリ内のCEL_filesフォルダに保存する。
【課題】
以下の手順に従って、マイクロアレイデータの正規化を行いなさい。
-
① ライブラリの準備
- Affymetrix 1色マイクロアレイデータを扱うために必要な R パッケージを読み込むこと。
- 正規化後の結果をExcelファイルとして出力できるようにすること。
-
② データの読み込み
- CEL_files フォルダ内に保存された.CELファイルを読み込み、Affymetrix 1色マイクロアレイの生データとしてRに取り込むこと。
- 作業ディレクトリを変更せず、パス指定で読み込むこと。
-
③ 正規化の実行
- 読み込んだ生データに対して、RMA (Robust Multi-array Average) 前処理を実行しなさい。この処理では、以下を一括で行うこと。
- 背景補正
- 正規化
- プローブレベルから遺伝子発現量への要約
- 読み込んだ生データに対して、RMA (Robust Multi-array Average) 前処理を実行しなさい。この処理では、以下を一括で行うこと。
-
④ 正規化後データの確認
- 正規化後の発現量データを用いて、サンプル間の分布をboxplotにより可視化しなさい。
- 正規化前後で分布が揃うことを確認しなさい。
-
⑤ 結果の出力
- 正規化後の遺伝子発現量行列を Excel ファイルとして出力しなさい。
- 出力ファイルは デスクトップに保存し、ファイル名は内容が分かるものとすること。
- 行名 (プローブ ID / 遺伝子 ID) を含めて出力すること。
解答
# =========================================
# DNAマイクロアレイ解析:正規化
# =========================================
# =========================================
# 0. ライブラリ
# =========================================
suppressPackageStartupMessages({
library(affy) # Affymetrix(1色)
library(limma) # 正規化・2色マイクロアレイ
library(openxlsx) # Excel 出力
})
# =========================================
# 1. 設定
# =========================================
array_type <- "one" # "one" = 1色(Affymetrix), "two" = 2色
data_dir <- "./CEL_files" # 1色マイクロアレイ用
desktop_path <- file.path(Sys.getenv("USERPROFILE"), "Desktop")
# =========================================
# 2. データ読み込み
# =========================================
if (array_type == "one") {
message("Reading CEL files for one-color microarray (Affymetrix)")
# 作業ディレクトリを変更せず、パス指定で読み込み
raw_data <- ReadAffy(celfile.path = data_dir)
} else if (array_type == "two") {
message("Reading raw data for two-color microarray")
# targets.txt と raw data が必要
targets <- readTargets("targets.txt")
# プラットフォームに応じて source を指定
RG <- read.maimages(targets, source = "agilent")
} else {
stop("array_type must be 'one' or 'two'")
}
# =========================================
# 3. 正規化
# =========================================
if (array_type == "one") {
message("Running RMA normalization for one-color microarray")
# ------------------------------------------------------------
# RMA 前処理
# (背景補正・正規化・要約を一括)
# ------------------------------------------------------------
eset <- rma(raw_data)
exprs_norm <- exprs(eset)
# 正規化後の分布確認
boxplot(exprs_norm,
main = "After RMA normalization",
outline = FALSE,
las = 2)
} else {
message("Running normalization for two-color microarray")
# ------------------------------------------------------------
# 色素内正規化(LOWESS)
# ------------------------------------------------------------
RG <- normalizeWithinArrays(RG, method = "loess")
# ------------------------------------------------------------
# アレイ間正規化
# ------------------------------------------------------------
RG <- normalizeBetweenArrays(RG, method = "Aquantile")
# 出力用データ
M <- RG$M # log2 比
A <- RG$A # 平均強度
}
# =========================================
# 4. 出力(Excel)
# =========================================
if (array_type == "one") {
out_path <- file.path(
desktop_path,
"Microarray_expression_RMA.xlsx"
)
write.xlsx(
as.data.frame(exprs_norm),
file = out_path,
rowNames = TRUE
)
} else {
out_path <- file.path(
desktop_path,
"Microarray_expression_twocolor.xlsx"
)
write.xlsx(
list(
M_value = as.data.frame(M),
A_value = as.data.frame(A)
),
file = out_path,
rowNames = TRUE
)
}
message("Excel file successfully exported to Desktop.")解説
5.1 ライブラリ
suppressPackageStartupMessages({
library(affy) # Affymetrix (1色)
library(limma) # 正規化・2色マイクロアレイ
library(openxlsx) # Excel 出力
})本スクリプトでは、DNAマイクロアレイデータの正規化および結果の出力を行うために、主に上記の3つのパッケージを利用しています。
-
affy パッケージ
Affymetrix社の1色マイクロアレイ (GeneChip) データを扱うための基本パッケージです。
affy は、CELファイルとして提供される Affymetrix の生データを読み込み、
- 背景補正
- 正規化
- プローブレベルから遺伝子レベルへの要約
といった前処理を一特に、RMA (Robust Multi-array Average) 法 が実装されており、Affymetrix マイクロアレイ解析では事実上の標準手法として広く用いられています。括で実行するための機能を備えています。
-
limma パッケージ
limmaはもともと差次的発現解析のために開発されましたが、現在では、
- 1色マイクロアレイの正規化
- 2色マイクロアレイの色素内正規化 (LOWESS)
- アレイ間正規化 (quantile、Aquantileなど)
といった前処理機能も広く利用されています。
特に2色マイクロアレイでは、RGListオブジェクトを用いたLOWESS正規化が標準的な解析手順として確立されています。
5.2 設定
# =========================================
# 1. 設定
# =========================================
array_type <- "one" # "one" = 1色(Affymetrix), "two" = 2色
data_dir <- "./CEL_files" # 1色マイクロアレイ用
desktop_path <- file.path(Sys.getenv("USERPROFILE"), "Desktop")
このセクションでは、解析対象となるマイクロアレイの種類や、データの保存場所、結果の出力先をあらかじめ指定しています。
array_type <- "one"この変数は、解析するマイクロアレイの種類を切り替えるための設定項目です。
- one: 1色マイクロアレイ (主にAffymetrix GeneChip)
- two: 2色マイクロアレイ (Agilentなど)
ここで指定した値に応じて、後続のスクリプトでは
読み込み方法や正規化手法が自動的に切り替わる構造になっています。
data_dir <- "./CEL_files"1色マイクロアレイ (Affymetrix) の生データ (CELファイル) が保存されているディレクトリを指定しています。
この行では、
- 作業ディレクトリ内の「CEL_files」フォルダの中を自動的に探し
- 拡張子”.CEL” (または .cel) のファイルをすべて読み込む
という動作を行っています。
Affymetrix データでは、フォルダごと複数のCELファイルをまとめて読み込むことが一般的であり、この変数によって、解析対象となるデータの場所を一元管理できるようになっています。
5.3 データ読み込み
if (array_type == "one") {
message("Reading CEL files for one-color microarray (Affymetrix)")
# 作業ディレクトリを変更せず、パス指定で読み込み
raw_data <- ReadAffy(celfile.path = data_dir)
} else if (array_type == "two") {
message("Reading raw data for two-color microarray")
# targets.txt と raw data が必要
targets <- readTargets("targets.txt")
# プラットフォームに応じて source を指定
RG <- read.maimages(targets, source = "agilent")
} else {
stop("array_type must be 'one' or 'two'")
}このセクションでは、マイクロアレイの種類に応じて、生データの読み込み方法を切り替えています。
前の設定で指定したarray_typeの値に基づき、1色マイクロアレイと2色マイクロアレイを分岐して処理します。
if (array_type == "one") {
message("Reading CEL files for one-color microarray (Affymetrix)")こでは、”array_type”が"one"に設定されている場合 (1色マイクロアレイを解析対象としている場合) の処理に入ります。
raw_data <- ReadAffy(celfile.path = data_dir)affy パッケージの ReadAffy() 関数を用いて、指定したディレクトリ (data_dir) 内にある CELファイルをまとめて読み込みます。
読み込まれたデータは、背景補正や正規化を行う前の生データ (raw data) としてraw_dataに格納されます。
} else if (array_type == "two") {“array_type”が"two"の場合、2色マイクロアレイ (Agilent など) のデータを読み込む処理に切り替わります。
targets <- readTargets("targets.txt")2色マイクロアレイでは、どのファイルがどのサンプル・色素に対応しているかを記述したtargets ファイルを用いるのが一般的です。
この部分では、targets.txtを読み込み、ファイル構成や実験条件の情報をRに取り込んでいます。
5.4 正規化
# =========================================
# 3. 正規化
# =========================================
if (array_type == "one") {
message("Running RMA normalization for one-color microarray")
# ------------------------------------------------------------
# RMA 前処理
# (背景補正・正規化・要約を一括)
# ------------------------------------------------------------
eset <- rma(raw_data)
exprs_norm <- exprs(eset)
# 正規化後の分布確認
boxplot(exprs_norm,
main = "After RMA normalization",
outline = FALSE,
las = 2)
} else {
message("Running normalization for two-color microarray")
# ------------------------------------------------------------
# 色素内正規化(LOWESS)
# ------------------------------------------------------------
RG <- normalizeWithinArrays(RG, method = "loess")
# ------------------------------------------------------------
# アレイ間正規化
# ------------------------------------------------------------
RG <- normalizeBetweenArrays(RG, method = "Aquantile")
# 出力用データ
M <- RG$M # log2 比
A <- RG$A # 平均強度
}このセクションでは、読み込んだ生データに対して正規化処理を行います。マイクロアレイでは、測定条件や技術的なばらつきの影響を受けやすいため、統計解析に進む前に、データの分布やスケールをそろえる処理が不可欠です。
ここでも、前段で指定したarray_typeに応じて、1色マイクロアレイと2色マイクロアレイで処理内容を切り替えています。
1色マイクロアレイの正規化
if (array_type == "one") {
message("Running RMA normalization for one-color microarray")“array_type“が"one"の場合、Affymetrixに代表される1色マイクロアレイの正規化を行います。
eset <- rma(raw_data)affyパッケージのrma() 関数を用いて、RMA前処理を実行します。
RMAでは、
- 背景補正
- アレイ間での分布の調整
- 複数プローブの信号をまとめた発現量の要約
これらの処理が一括で行われます。
exprs_norm <- exprs(eset)RMA 処理後の発現量データを取り出し、
行に遺伝子、列にサンプルを持つ発現行列としてexprs_normに格納します。
この行列が、以降の統計解析や可視化の基礎となります。
boxplot(exprs_norm,
main = "After RMA normalization",
outline = FALSE,
las = 2)正規化後のデータ分布を確認するため、各サンプルの発現量分布を箱ひげ図で表示しています。
サンプル間で分布の形が概ねそろっていれば、正規化が適切に機能していると判断できます。
2色マイクロアレイの正規化
} else {
message("Running normalization for two-color microarray")“array_type”が"two"の場合は、2色マイクロアレイの正規化処理に進みます。
RG <- normalizeWithinArrays(RG, method = "loess")同一アレイ内での色素間の偏りを補正します。
2色マイクロアレイでは、蛍光色素ごとの発光効率や強度依存性の違いにより、赤と緑の信号比が歪むことがあります。
ここでは loess 法を用いて、こうした色素由来の系統的な偏りを補正しています。
RG <- normalizeBetweenArrays(RG, method = "Aquantile")次に、アレイ間のばらつきを補正します。複数のスライド間で強度分布をそろえることで、条件間比較がしやすいデータに整えています。
M <- RG$M
A <- RG$A正規化後のデータから、
- M値: log2 スケールでの発現比
- A値: 2色の平均強度
を取り出し、出力や後続解析に利用できる形にしています。
このように、正規化の段階でマイクロアレイの測定原理に応じた補正を行うことで、技術的なばらつきを抑えた、解釈しやすいデータが得られます。
まとめ
本稿では、「DNAマイクロアレイデータの正規化」を主題として、DNAマイクロアレイ解析の基礎概念から、データに内在するばらつきの種類、正規化手法の考え方、そしてRを用いた実践的な正規化処理までを一連の流れとして解説しました。
DNAマイクロアレイ解析は、RNA を手がかりに「どの遺伝子が、どの程度発現しているか」を網羅的に把握するために開発され、トランスクリプトミクス研究の基盤を築いてきた手法です。マイクロアレイには、1色マイクロアレイと2色マイクロアレイという異なる測定方式が存在し、それぞれで得られるデータ構造や主要なバイアスの性質が異なります。
また、マイクロアレイデータには、遺伝子発現そのものとは無関係な技術的・測定的ばらつきが必ず含まれます。これらを補正せずに解析を進めると、遺伝子発現の差を過大または過小に評価し、誤った解釈につながる可能性があります。そのため、正規化はマイクロアレイ解析における不可欠な前処理工程です。
本稿では、
- 全体強度の正規化 (Global normalization)
- 中央値正規化 (Median normalization)
- LOWESS正規化
- 分位点正規化 (Quantile normalization)
といった代表的な正規化手法を整理し、それぞれがどのようなバイアスを補正するための方法であるかを確認しました。特に、2色マイクロアレイでは色素間バイアスや強度依存性バイアスの補正が中心となり、1色マイクロアレイではアレイ間の分布を揃えることが重視される点が、解析設計上の重要な違いとなります。
DNAマイクロアレイ解析は、単に正規化手法を機械的に適用すればよいものではありません。「どのようなバイアスが存在し、何を補正しているのか」を理解したうえで解析を進めることが、結果を正しく解釈するための鍵となります。
RNA-seqが主流となった現在においても、DNAマイクロアレイは、既存データ資産の活用や条件間比較を目的とした解析において、依然として有用な手法です。本稿が、DNAマイクロアレイ解析を改めて体系的に理解し、RNA-seqとの違いや使い分けを自ら説明できるようになるための一助となれば幸いです。
関連するサービス

参考文献
- 辻本豪三. 網羅的遺伝子発現解析 (DNAチップ,マイクロアレイ) とそのゲノム創薬への応用. 日本薬理学雑誌. 2005;125(3):153–157. doi: 10.1254/fpj.125.153.
- 中井雄治. ニュートリゲノミクス研究で培われたDNAマイクロアレイ解析パイプラインの他分野への応用. 日本生化学会誌. 2016;88(1):7–14. doi: 10.14952/SEIKAGAKU.2016.880007.
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002 Jan 1;30(1):207-10. doi: 10.1093/nar/30.1.207. PMID: 11752295; PMCID: PMC99122.
- Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Rocca-Serra P, Sansone SA. ArrayExpress--a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003 Jan 1;31(1):68-71. doi: 10.1093/nar/gkg091. PMID: 12519949; PMCID: PMC165538.
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002 Dec;32 Suppl:496-501. doi: 10.1038/ng1032. PMID: 12454644.
- Fujita A, Sato JR, Rodrigues Lde O, Ferreira CE, Sogayar MC. Evaluating different methods of microarray data normalization. BMC Bioinformatics. 2006 Oct 23;7:469. doi: 10.1186/1471-2105-7-469. PMID: 17059609; PMCID: PMC1636075.
- Microarray Data Analysis for Genetic Network Prediction. Retrieved January 15, 2026, from https://yishi.sjtu.edu.cn/microarray/survey_normalization.htm
- Dobbin KK, Kawasaki ES, Petersen DW, Simon RM. Characterizing dye bias in microarray experiments. Bioinformatics. 2005 May 15;21(10):2430-7. doi: 10.1093/bioinformatics/bti378. Epub 2005 Mar 17. PMID: 15774555.
- Barnes M, Freudenberg J, Thompson S, Aronow B, Pavlidis P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res. 2005 Oct 19;33(18):5914-23. doi: 10.1093/nar/gki890. PMID: 16237126; PMCID: PMC1258170.
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004 Feb 12;20(3):307-15. doi: 10.1093/bioinformatics/btg405. PMID: 14960456.
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015 Apr 20;43(7):e47. doi: 10.1093/nar/gkv007. Epub 2015 Jan 20. PMID: 25605792; PMCID: PMC4402510.
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002 Feb 15;30(4):e15. doi: 10.1093/nar/30.4.e15. PMID: 11842121; PMCID: PMC100354.
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, Gottardo R, Hahne F, Hansen KD, Irizarry RA, Lawrence M, Love MI, MacDonald J, Obenchain V, Oleś AK, Pagès H, Reyes A, Shannon P, Smyth GK, Tenenbaum D, Waldron L, Morgan M. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015 Feb;12(2):115-21. doi: 10.1038/nmeth.3252. PMID: 25633503; PMCID: PMC4509590.