RNA-seq解析をブラックボックスにしないために

- アウトライン
-
- 作成日: 2025/12/12
- 更新日: –
はじめに
生物の設計図とも例えられるDNAには、細胞のふるまいを決定する多くの情報が記録されています。その中には、タンパク質をつくるための遺伝子配列も含まれており、細胞は状況に応じて、これらの遺伝子の情報を 「使うか/使わないか」 を精密に調節しています。
このように、遺伝子の情報が実際に利用され、遺伝子産物の合成に用いられる一連の過程を 遺伝子発現 と呼びます。遺伝子が発現する際、DNAに記録された情報は、まず RNA へと写し取られます。この過程を 転写 (transcription) といいます。
転写によって産生されるRNAにはさまざまな種類がありますが、そのうちタンパク質へと翻訳されるものが mRNA (messenger RNA) です。細胞は環境や生理的状態に応じて、どの遺伝子をどの程度転写するかを選択的に制御しているため、RNAの量を調べることで、遺伝子発現の状態を把握することができます。
このような転写産物 (RNA) の量を網羅的に測定する代表的な手法が RNA-seq (RNA sequencing) です。RNA-seqでは、細胞内に存在するRNAを包括的に読み取り、その発現量を定量することで、細胞の状態や刺激に対する反応の変化を詳細に解析することが可能となります。
問題を始める前に
2.1 RNA-seqとは?
RNA-seq (RNAシーケンス) は、次世代シーケンサー (NGS) を用いて、細胞や組織から抽出したRNAを網羅的に読み取り、その配列情報と量を解析する手法です。これにより、細胞が どの遺伝子を、どの程度利用しているのか を高い解像度で把握することができます。
従来の遺伝子発現解析法 (マイクロアレイなど) では、あらかじめ設計されたプローブ配列と相補的な遺伝子のみを対象として発現量を測定していました。一方、RNA-seqでは、細胞内に存在する多様なRNAを高感度に検出することが可能であり、既知の遺伝子に加えて未知の転写産物や新規スプライシング変異の解析にも適しています。
また、発現量が極めて低い遺伝子や、特定の条件下でのみ変動する遺伝子についても定量性高く捉えることができるため、条件間のわずかな発現差を検出することが可能です。
このような高い網羅性と精密性を支えているのが次世代シーケンサー (NGS) です。NGSは、従来のサンガー法による配列決定とは異なり、膨大な数のDNA断片を同時並行的に解析することができます。その結果、一度の解析で大量のリード(配列情報)を取得することができ、複数サンプルの同時解析や、環境刺激・疾患・薬剤処理などに伴う遺伝子発現の時間的変化を追跡することも容易になります。
RNA-seqから得られる膨大な配列データは、単にRNAの存在を記録するだけでなく、「どの遺伝子が、どの条件下で、どの程度変化しているのか」を明らかにするための強力な情報源となります。特に、条件や群間の違いに着目した遺伝子発現変動の解析は、生体反応の理解、病態メカニズムの解明、さらにはバイオマーカー探索などへと直結する重要なアプローチです。
2.2 差次的発現解析とは?
2つ以上の条件 (処理群とコントロール群など) の間で、「どの遺伝子の発現量が統計的に有意に変化しているか」 を調べる解析を、差次的発現解析 (differential expression analysis) と呼びます。
RNA-seqによって得られる遺伝子ごとのカウントデータをもとに、統計モデルを用いて条件間の発現量の差を検定することで、どの遺伝子が条件の違いに応じて有意に変動しているかを明らかにします。
差次的発現解析では、一般に以下の工程を経て、条件間の差を反映して変動している遺伝子を抽出します。
- 正規化 (Normalization)
サンプル間で読み取られた総リード数の違いや、全体的な発現量分布の偏りを補正し、遺伝子発現量を相互に比較可能な状態に整えます。 - 統計モデル化
RNA-seqのカウントデータは分散が大きく、平均と分散の関係が単純ではないため、その特性を適切に表現できる 負の二項分布 などの統計モデルを用いて解析を行います。 - 条件間の比較検定
各遺伝子について、発現量が条件間 (群間) で有意に異なるかどうかを統計的に検定します。 - 多重比較補正 (False Discovery Rate; FDR)
数千から数万の遺伝子を同時に検定するため、多重検定による偽陽性の増加を抑える目的で、False Discovery Rate (FDR) などを用いた補正を行います。
2.3 DEGsとは?
差次的発現解析の結果、条件間で統計的に有意な発現変動 (増加または減少) を示した遺伝子は、差次的発現遺伝子 (Differentially Expressed Genes; DEGs) と呼ばれます。
DEGsの判定には、主に以下の指標が用いられます。
- log2 Fold Change (log2FC)
条件間における発現量の変化の大きさを表す指標であり、正の値は発現の増加、負の値は発現の減少を示します。 - 調整済み p 値 (adjusted p-value)
多数の遺伝子を同時に検定することによる多重検定の影響を補正した p 値で一般に False Discovery Rate (FDR) に基づく補正が用いられ、padjとして表記されることが多いです。
これらの指標をもとに、例えば padj<0.05などの基準を満たす遺伝子を、差次的発現が認められた遺伝子、すなわち DEGsとして取り扱います。
問題
RNA-seqは、細胞内に存在するRNAを網羅的に測定し、条件間での遺伝子発現の違いを解析するための強力な手法である。本問題では、Rに用意されている公開RNA-seqデータセットを用いて、処理群とコントロール群の間で差次的に発現している遺伝子 (DEGs) を同定し、その結果を可視化してください。
【データ】
- Bioconductor パッケージ airway に含まれる RNA-seq データを使用する
- 本データは、デキサメタゾン (dex) 処理の有無による遺伝子発現の違いを測定したものである
- dex = trt: 処理群
- dex = untrt: コントロール群
【課題】
RNA-seq のカウントデータおよび各サンプルの処理条件 (dex) を読み込み、遺伝子 × サンプルのカウント行列とサンプル情報を確認した上で、DESeq2を用いて差次的発現解析を行ってください。解析にあたっては、デザイン式に処理条件 (dex) を用い、コントロール群 (untrt) を基準としてモデルを構築してください。次に、多重比較補正後のp値 (False Discovery Rate; FDR) を用いて、padj < 0.05を満たす遺伝子を差次的発現遺伝子 (DEGs) として抽出してください。さらに、得られた解析結果をもとに、PCAプロットを作成して群間の発現パターンの違いを確認するとともに、DEGsの上位遺伝子を用いたヒートマップおよびVolcano plotを作成し、遺伝子発現の変動量と統計的有意性の関係を可視化してください。
解答
# サンプルデータの読み込み
library(airway)
data("airway")
# カウント行列(遺伝子 × サンプル)
count_data <- assay(airway)
# サンプル情報(投与 / 非投与)
col_data <- as.data.frame(colData(airway))
col_data$dex <- relevel(col_data$dex, "untrt")
head(count_data[, 1:3])
head(col_data)
# DESeq2 のモデル構築
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = count_data,
colData = col_data,
design = ~ dex)
dds <- DESeq(dds)
res <- results(dds)
head(res)
# 有意な差次的発現遺伝子(DEGs)の抽出
# 5% FDRで抽出
sig <- res[res$padj < 0.05 & !is.na(res$padj), ]
head(sig[order(sig$log2FoldChange, decreasing = TRUE), ])
# PCAプロット(群間の違いが視覚化される)
vsd <- vst(dds)
plotPCA(vsd, intgroup="dex")
# ヒートマップ
library(pheatmap)
top_genes <- rownames(sig)[1:50]
annotation_col <- data.frame(
dex = colData(vsd)$dex
)
rownames(annotation_col) <- rownames(colData(vsd))
ann_colors <- list(
dex = c("trt" = "orange", "untrt" = "green")
)
pheatmap(
assay(vsd)[top_genes, ],
show_rownames = FALSE,
annotation_col = annotation_col,
annotation_colors = ann_colors,
annotation_names_col = FALSE
)
# Volcano plot
library(EnhancedVolcano)
EnhancedVolcano(res,
lab = rownames(res),
x = 'log2FoldChange',
y = 'pvalue',
pCutoff = 0.05)
解説
5.1 library(airway)
library(airway)Bioconductorが提供するRNA-seqのサンプルデータセット「airway」を利用するために、対応するパッケージを読み込むコマンドです。
airwayは、
- デキサメタゾン (dexamethasone) 処置群 (trt)
- 非処置群 (untrt)
の2群から構成されるRNA-seq 用の公開データセットです。
このデータセットは、RangedSummarizedExperimentクラスのオブジェクトとして提供されており、遺伝子ごとの数値データ (例: RNA-seq のカウント) と、それに対応するゲノム情報やサンプル情報をひとつのオブジェクトにまとめて扱えるよう設計されています。
5.2 data("airway")
data("airway")airwayオブジェクトをメモリ上にロードするコマンドです。
読み込まれたairwayはSummarizedExperimentクラス (正確には RangedSummarizedExperiment) に属するオブジェクトであり、RNA-seq解析に必要な複数の情報が統合的に格納されています。
具体的には、以下の要素が含まれています。
- assay()
遺伝子 × サンプルのカウントデータ (RNA-seq の read counts) - colData()
サンプル情報 (処置条件、細胞株など) - rowData()
遺伝子情報 (遺伝子 ID、遺伝子長、ゲノム座標など
5.3 カウント行列を取り出す
count_data <- assay(airway)airway オブジェクトの assay (実験データ本体) から、RNA-seqのカウント行列を抽出します。
このカウント行列は、以下のような構造を持ちます。
| 遺伝子 | Sample1 | Sample2 | Sample3 | ... |
|---|---|---|---|---|
| geneA | 120 | 98 | 200 | ... |
| geneB | 5 | 8 | 2 | ... |
これは、各遺伝子が各サンプルで何回読み取られたか (read counts) を表す RNA-seqの生データであり、差次的発現解析などの統計解析の入力データとして用いられます。
5.4 サンプル情報を取り出す
col_data <- as.data.frame(colData(airway))airway オブジェクトに含まれるサンプル情報 (metadata) を取得し、data.frame として扱いやすい形式に変換します。
例えば、以下のような情報が含まれています。
| sample | dex(処置) | cell(細胞株) |
|---|---|---|
| SRR…1 | untrt | N61311 |
| SRR…2 | trt | N61311 |
| SRR…3 | untrt | N052611 |
| SRR…4 | trt | N052611 |
このデータフレームには、各サンプルに対応する 実験条件や群情報 が格納されており、後続の差次的発現解析において重要な役割を果たします。
5.5 因子レベルの基準 (ref) を “untrt” に設定
col_data$dex <- relevel(col_data$dex, "untrt")RNA-seq 解析では、処置条件 (ここではdex) を 因子 (factor) として扱い、そのうえで どの水準を基準 (reference level) とするか を明示的に指定します。上記のコマンドでは、非処置群(untrt) を基準レベルとして設定しています。
この操作が重要である理由は、DESeq2 における線形モデルが、基準レベルとの差として遺伝子発現の変化を推定する仕組みになっているためです。具体的には、log2 Fold Change は次のように定義されます。
すなわち、基準として設定したuntrtを0としたときに、処置群 (trt) でどの程度発現量が変化したかを推定しています。
このように基準を untrt に設定しておくことで、
- untrt: コントロール(基準)
- trt: 処置の効果 (treatment effect)
という形でlog2FoldChangeを直感的に解釈することができます。
その結果、後から得られる DEGs(差次的発現遺伝子) についても、「処置によって発現が増加した遺伝子か、減少した遺伝子か」という 変動の方向性 を分かりやすく判断できるようになります。
5.6 データの確認
head(count_data[, 1:3])
head(col_data)上記のコマンドにより、count_dataおよびcol_dataの先頭数行を確認し、サンプルのラベルや条件設定に誤りがないかをチェックします。この工程は、解析前の品質管理 (Quality Control; QC) の一環として重要です。
● count_dataの確認
遺伝子名の並び方や、リード数のおおよそのスケール、ゼロが極端に多くないかについて確認を行っています。
● サンプル情報の最初の行を見る
群分けが正しく untrt / trt の2条件になっているか、細胞株 (cell line) が2種類含まれていることは想定どおりかを確認しています。
これらを確認することで、後続の差次的発現解析におけるトラブルや解釈ミスを未然に防ぐことができます。
5.7 DESeq2のモデル構築
library(DESeq2)library(DESeq2)により、Bioconductorが提供するDESeq2パッケージを読み込みます。DESeq2は、RNA-seqのraw countデータを統計モデルに基づいて解析し、処理条件の違いによって発現が変化する遺伝子を検出するための代表的な手法です。
dds <- DESeqDataSetFromMatrix(
countData = count_data,
colData = col_data,
design = ~ dex)このコマンドでは、DESeq2 による解析に必要な情報を指定しています。
- countData
各遺伝子 × 各サンプルのrawカウントデータ (整数値) - colData
サンプル情報 (処置条件などのメタデータ) - design = ~ dex
「何を要因としてサンプル間を比較するか」を指定するデザイン式
ここでは、デキサメタゾン処理の有無 (dex) による遺伝子発現の違いを評価することを意味します。
dds <- DESeq(dds)DESeq() は解析の中心となる関数であり、以下の処理を自動的に実行します。
- カウントデータの正規化 (ライブラリサイズ補正)
- 分散推定 (遺伝子ごとのばらつきをモデル化)
- 負の二項分布モデルによる発現差の推定
- Wald 検定による統計的有意性の評価
res <- results(dds)
head(res)このコマンドにより、群間比較の結果をデータフレーム形式で取得できます。resには、各遺伝子について以下の情報が含まれています。
- log2 Fold Change
発現量の変化の大きさ(2 倍増加で +1、半分で −1) - pvalue
検定によるp値 - padj (FDR)
多重検定補正後のp値 (調整済みp値)
head()を用いて、解析結果の構造や値の範囲を確認しています。ここで得られた結果をもとに、有意に変動している遺伝子を抽出し、後続の解析や生物学的解釈へと進みます。
5.8 DESeq2 による差次的発現解析の実行
差次的発現解析によって遺伝子ごとの発現差を推定した後、どの遺伝子が統計的に有意に変動したか を判断するために、DEGs (差次的発現遺伝子) の抽出を行います。
sig <- res[res$padj < 0.05 & !is.na(res$padj), ]この処理では、DESeq2 が出力した結果 res の中から、FDR < 0.05(5%) を満たす遺伝子のみを抽出しています。
- res$padj < 0.05
多重検定補正後のp値 (調整p値) が 0.05 未満の遺伝子を選択 - !is.na(res$padj)
padj が NA (低カウントなどにより統計量が計算できない遺伝子) を除外
これにより、多重検定の影響を考慮したうえで、発現変動が生じている可能性が高い遺伝子 を抽出できます。
head(sig[order(sig$log2FoldChange, decreasing = TRUE), ])抽出したDEGsを、log2 Fold Change (発現変化量) が大きい順に並べ替えて確認しています。
- order(sig$log2FoldChange, decreasing = TRUE)
正方向に強く発現が増加した遺伝子から順に並び替え
この操作により、処理によって特に大きく誘導された遺伝子を直感的に把握することができます。
5.9 PCA プロットの描画
以下のコマンドを用いて、主成分分析 (Principal Component Analysis; PCA) を行います。
vsd <- vst(dds)RNA-seqデータでは、発現量の大きい遺伝子ほど分散も大きくなるという特性があり、生のカウントデータのままPCAを行うと、一部の高発現遺伝子に解析結果が強く影響されてしまう という問題があります。
この問題を解消するために、分散安定化変換 (variance stabilizing transformation; vst) を適用します。
vstを行うことで、主に以下の処理が行われます。
- カウントデータを連続値に変換
- 発現量の大小に依存せず、分散がほぼ一定になるように補正
- PCAやクラスタリングに適したスケールへ変換
plotPCA(vsd, intgroup="dex")vsdに対してplotPCA()を実行することで、PCA プロットを作成します。
- intgroup = "dex"
処置条件 (dex:untrt / trt) を群として指定し、サンプルを色分けして表示します。
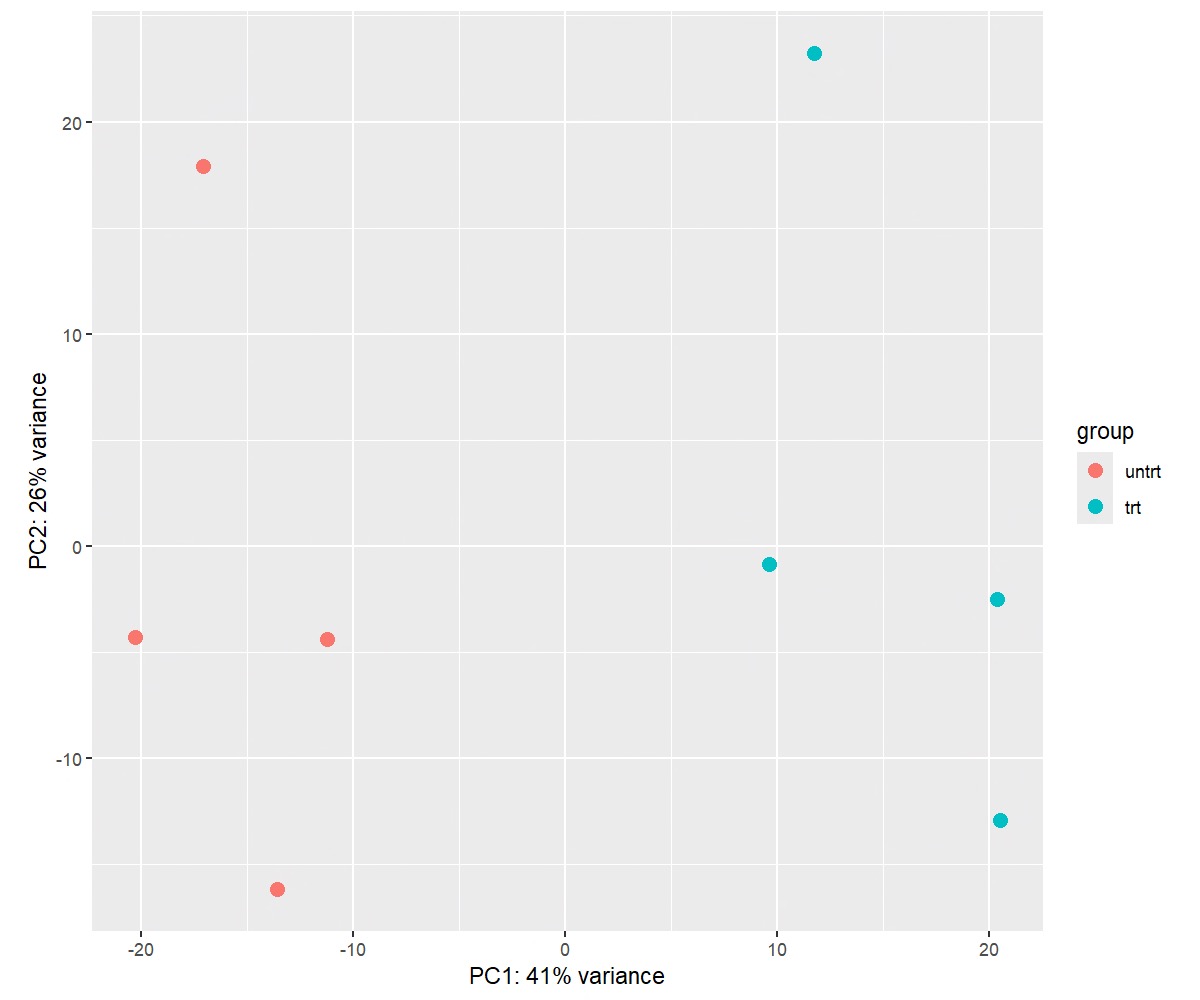
図1: PCAプロット
RNA-seq 解析では、数万に及ぶ遺伝子の発現量が同時に測定されるため、サンプル間の違いを数値だけから直感的に把握することは困難です。そこで有用なのが、このPCAプロット です。
PCAプロットでは、各サンプルが持つ膨大な遺伝子発現情報を2次元に要約し、サンプル間の類似度を距離として表現することで、地図のように可視化します。
今回の結果では、
- 未処理群 (untrt)
- デキサメタゾン処理群 (trt)
の2群が、それぞれまとまって明確に分離していることが確認できます。このことから、デキサメタゾン処理によって遺伝子発現プロファイルが大きく変化している ことが、PCA プロットから一目で読み取れます。
5.10 ヒートマップの描画
library(pheatmap)pheatmapパッケージを読み込みます。
これは、遺伝子発現データの ヒートマップ描画 に広く用いられているパッケージです。
top_genes <- rownames(sig)[1:50]発現変化量 (log2 Fold Change) が大きい順に並び替えた有意なDEGsの一覧であるsigの中から、上位50遺伝子を抽出しています。
annotation_col <- data.frame(
dex = colData(vsd)$dex
)
rownames(annotation_col) <- rownames(colData(vsd))vsdに対応するサンプル情報から、処置条件であるdexを取り出し、pheatmap に渡すための 列注釈 (annotation_col) を作成しています。
この注釈は、「どのサンプルがどの群に属するか」をヒートマップ上部に色帯として表示するために用いられます。
ann_colors <- list(
dex = c("trt" = "orange", "untrt" = "green")
)処置群 (trt) と非処置群 (untrt) の色分けを指定しています。
pheatmap(
assay(vsd)[top_genes, ],
show_rownames = FALSE,
annotation_col = annotation_col,
annotation_colors = ann_colors,
annotation_names_col = FALSE
)pheatmap() を用いて、ヒートマップを描画します。
各引数の意味は以下のとおりです。
- assay(vsd)[top_genes, ]
vst 変換後の正規化データ (vsd) から、行方向に上位 50 の DEGs を抽出して使用します。 - show_rownames = FALSE
遺伝子名が多くなり図が読みにくくなるため、行名を非表示にしています。 - annotation_col = annotation_col
サンプルの処置条件 (dex) を、ヒートマップ上部に色帯として表示します。 - annotation_colors = ann_colors
trt / untrt に対応する色指定を適用します。 - annotation_names_col = FALSE
注釈名を非表示にし、図を簡潔にしています。

図2: ヒートマップ
ヒートマップは、差次的発現遺伝子 (DEGs) の中でも特に変動の大きい遺伝子を並べ、その発現レベルを色の違いとして可視化する手法です。
縦軸に遺伝子、横軸にサンプルを配置し、色の濃淡によって「どの程度発現しているか」を表現します。
今回のヒートマップでは、
- 処置群 (trt) と非処置群 (untrt) で、明確に異なる色パターン が観察され、デキサメタゾン処理による遺伝子発現の変化が視覚的に確認できます。
- クラスタリング (左側および上部の樹形図) により、発現パターンが類似した遺伝子同士、ならびにサンプル同士が自動的にグループ化されています。
5.11 ボルケーノプロットの描画
RNA-seq解析の結果として得られたDEGs (差次的発現遺伝子) を視覚的に把握するための代表的な図が、ボルケーノプロット (volcano plot) です。
ボルケーノプロットでは、以下の2つの情報を同時に確認することができます。
- 遺伝子の発現変動量 (効果の大きさ: Fold Change)
- その変動の統計的信頼性 (p値)
library(EnhancedVolcano)EnhancedVolcanoパッケージを読み込みます。
このパッケージは、RNA-seqの差次的発現解析結果を分かりやすくボルケーノプロットとして可視化するために用いられます。
EnhancedVolcano(res,
lab = rownames(res),
x = 'log2FoldChange',
y = 'pvalue',
pCutoff = 0.05)
このコマンドにより、DESeq2 の解析結果を用いたボルケーノプロットを作成します。
各引数の意味は以下のとおりです。
- EnhancedVolcano(res, ...)
DESeq2 の結果オブジェクト res(log2FoldChange、p 値、padj などを含む)を指定します。 - lab = rownames(res)
プロット上に表示する遺伝子名(ラベル)を指定します。
ここでは、res の行名に対応する遺伝子名を用いています。 - x = "log2FoldChange"
- 横軸に log2 Fold Change (発現変動量) を表示します。正の値は 発現上昇 (up-regulated)、負の値は 発現低下 (down-regulated) を示します。火山図における左右方向の広がりが、発現変化の大きさを表しています。
- y = "pvalue"
縦軸に p 値を指定します。EnhancedVolcano では自動的に −log10 (p-value) に変換して描画されるため、上に位置する点ほど統計的有意性が高いことを意味します。 - pCutoff = 0.05
- 指定した p 値 (0.05) を基準として、点の色分けが行われます。
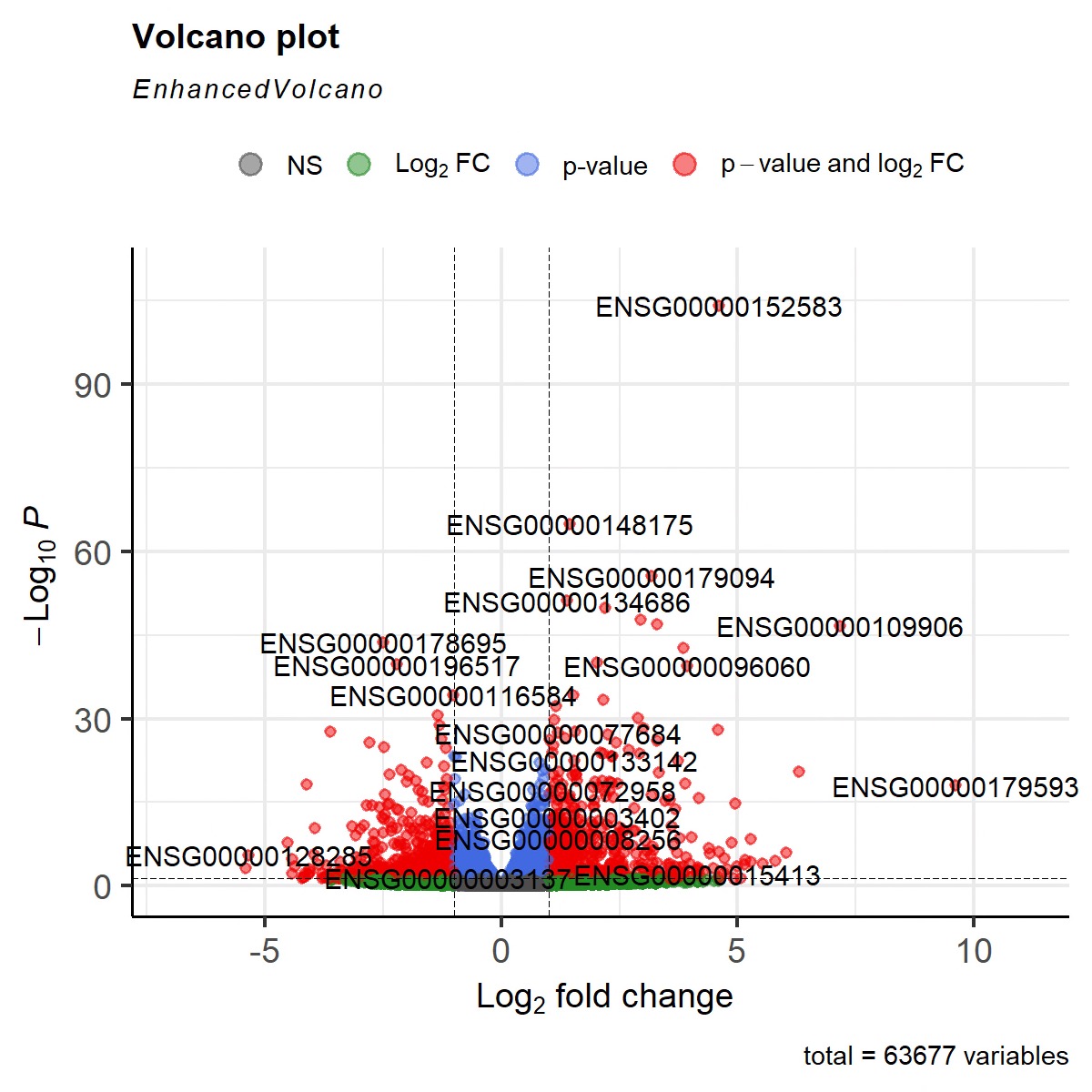
図3: ボルケーノプロット
ボルケーノプロットは、差次的発現解析の結果を発現変動量と統計的有意性の両面から要約した図です。
各遺伝子を、以下の 2 軸からなる散布図として表現します。
- 横軸:log2 Fold Change (どれだけ発現が変動したか)
- 縦軸:−log10 p-value (どれだけ統計的に確からしい差か)
この図から、次のような解釈が可能です。
- 右側に大きく位置する点: 処理によって発現が上昇した遺伝子
- 左側に大きく位置する点: 発現が低下した遺伝子
- 上方に位置する点: p値が小さく、統計的有意性が高い遺伝子
まとめ
本稿は、「RNA-seq解析をブラックボックスにしない」ことを目的として、RNA-seqの基礎概念から差次的発現解析、さらに R(DESeq2)を用いた実践的な解析手順までを一連の流れとして解説しました。
RNA-seq は、次世代シーケンサー (NGS) を用いてRNAを網羅的に測定することで、細胞が どの遺伝子を、どの程度利用しているのかを高い解像度で捉えることができる手法です。しかし実際の解析では、
「なぜこの前処理が必要なのか」
「この数値は何を意味しているのか」
といった部分が曖昧なまま、結果だけを受け取ってしまうケースも少なくありません。
そこで本稿では、差次的発現解析の考え方を正規化・統計モデル・多重検定補正というステップに分解し、DEGs(差次的発現遺伝子)がどのような根拠で同定されているのかを丁寧に整理しました。また、DESeq2 を用いた解析例を通じて、
- log2 Fold Changeの意味
- p 値・FDR (padj) の役割
- 基準群 (reference level) 設定の重要性
といった、結果解釈に不可欠なポイントを具体的に確認しました。
さらに、PCA プロット・ヒートマップ・ボルケーノプロットといった代表的な可視化手法を用いることで、「どの遺伝子が変わったか」だけでなく、「サンプル全体として何が起きているのか」を俯瞰的に理解できることも示しました。これらの図は、解析結果を第三者に説明する際や、生物学的な仮説を立てる際に非常に強力なツールとなります。
RNA-seq 解析は、コードを実行すれば結果が出るという意味では「簡単」に見えるかもしれません。しかし、その裏側には明確な統計的前提と設計思想があります。解析の各ステップの意味を理解することが、結果を正しく解釈し、研究の質を高める第一歩です。
本稿が、RNA-seq 解析を「よく分からないブラックボックス」ではなく、自分の言葉で説明できる解析手法として捉えるための一助となれば幸いです。
関連するサービス

参考文献
- 一般社団法人 日本臨床衛生検査技師会(監修). 遺伝子・染色体検査技術教本. 東京: 丸善出版.
- Love MI, Huber W, Anders S. Analyzing RNA-seq data with DESeq2.
Bioconductor Vignette [Internet]. Available from: https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html